신약개발 본질에서 멀어질수록 더 가까워지는 '코리아 패싱'
한국 신약이 한국을 떠나는 아이러니… '규제'기관은 '지원'도 해야 한다
THINK 한국의 신약개발을 어찌할 것인가
한 해가 꺾이는 6월을 떠나보냈다. 한겨울부터 초여름까지 <히트뉴스> 한 켠에 단단히 매달려 있던 문제의식은 '한국의 신약개발을 어찌할 것인가'다. 잊을 만하면 들려오는 신약 라이선싱 소식에 기뻐하다가도, 이런 소식을 왜 대부분의 바이오텍이 들을 수 없는지 고민했다. 한국에는 대체 무엇이 부족한 것인가?
[끝까지 HIT 10호] 국내외를 넘나들며 같은 질문을 반복하고 나니, 우리의 부족함은 3개로 명료하게 떨어진다. 인재가 부족하고, 돈이 부족하고, 정책이 부족하다. 어느 하나 쉬이 풀 수 없지만, 몇몇 기업들은 기민하게 움직여 어떻게든 인재를 구하고 투자를 유치한다. 그렇게 성과를 만들어 왔건만, 여전히 그들을 좌절하게 하는 건 결국 정책이다.
코로나19 당시 미국의 mRNA 백신은 놀라운 속도로 개발되고 상업화됐다. 그 배경에는 미국 식품의약국(FDA) 특유의 레퍼런스(Reference) 시스템이 있었다. FDA는 mRNA 백신 플랫폼이 이미 코로나19 전에 임상개발을 거쳤다는 점을 참고해, 약간의 변형이 가해진 코로나19 mRNA 백신의 개발 기간을 단축시켰다. 그렇게 화이자의 '코미나티(COMIRNATY)'는 2020년 12월에 긴급사용승인(EUA)을 거쳐 2021년 8월에 정식 승인을 받는 데 이르렀다.
반면 한국의 첫 국산 코로나19 백신이었던 '스카이코비원'은 2022년 6월에 식품의약품안전처의 품목허가를 받았다. 코미나티에 비해 약 4개월 늦게 개발이 시작됐음을 감안해도 허가 시기가 너무나 늦었다. 규제기관에 레퍼런스 시스템이 없어, 6개월의 추가 독성시험 기간을 거쳐야 했던 게 치명적이었다. 결국 외산 mRNA 백신들이 차지한 시장에 뒤늦게 진입한 토종 백신은 아직까지도 고전을 면치 못하고 있다.
코로나19 사태가 끄집어낸 한국 규제기관의 역량 문제는 비단 백신에만 국한된 것이 아닐 터다. 사실 이 문제는 많은 분야에서 되풀이돼 왔으며, 그 중 가장 아픈 손가락을 꼽자면 첨단재생의료를 말할 수밖에 없다. '인보사'ㆍ'조인트스템'의 상흔이 남은 언덕이다.
2019년 품목허가 취소를 당한 인보사는 5년이 넘은 지금까지도 시장에 돌아오지 못하고 있다. 사건이 오래된 만큼, 적잖은 이들은 인보사에 치명적인 효력 문제나 안전성 문제가 있었던 것으로 기억하고 있다. 그러나 인보사의 효력에는 의문의 여지가 없다는 게 규제 당국과 의료계의 공통된 반응이다. 문제는 뒤바뀐 세포주에 있었다.
인보사의 2액에는 본래 유전자 조작된 연골세포가 들어 있어야 했으나, 코오롱생명과학의 실수로 신장 유래 세포가 대신 들어가 있었다. 물론 신장 유래 세포가 발암성 물질일 수 있다는 식약처의 염려는 타당했으며, 이에 품목허가 취소 결정도 이해 가능한 수준의 조치라 할 수 있다.
하지만 5년에 걸친 법정 다툼으로 인해, 인보사의 과오를 바로잡고 환자들에게 신규 치료 옵션을 제공할 수 있는 기회는 점점 멀어져만 가고 있다. 규제기관 입장에서 세포주 문제를 빠르게 정리하고 인보사를 시장에 재도입시킬 수 있는 방법은 분명 있었을 텐데도 말이다.
결국 코오롱생명과학은 미국에서 살 길을 모색하기로 정한 듯하다. 세포주 착오에 대한 사측의 소명을 받아들인 FDA는 인보사의 미국 임상 3상을 재개시켰다. 추적관찰을 포함해 오는 2027년에 시험이 종료될 예정으로, 임상결과가 좋을 경우 미국 내 허가는 이듬해 이뤄질 것으로 보인다.
알바이오의 조인트스템 허가 불발 사건도 같은 맥락상에서 짚어야 하는 문제다. 무릎 퇴행성 관절염에 대한 세포치료제인 조인트스템은 작년 4월에 허가 신청이 반려됐다. 반려 사유는 2차 중앙약사심의위원회(중앙약심)가 제기한 '임상적 유의성 부족'이었다.

그러나 중앙약심은 사전에 정의된 기준을 두고 임상적 유의성을 평가하지 않았다. 심지어 이들이 사용한 기준은 알바이오가 시험 전에 예상해 둔 효력 점수차였다. 달리 빗대자면, 시험 출제자가 합격선 점수를 미리 정해두지 않고, 학생이 예상한 점수를 두고 합불을 정했다. '학생인 네가 100점을 받겠다 했는데, 90점을 받았으니 불합격시키겠다'고 말하는 것과 다름없다.
이를 두고 알바이오가 미국 2b/3a상 시험에 더욱 몰두하게 된 건 자연스러운 수순이다. 한국 규제기관의 역량 문제는 코오롱생명과학에 이어 알바이오까지 미국을 먼저 바라보게 만들었다. '국산 신약이 한국이 아닌 미국에서 먼저 출시될 수도 있겠다'는 걱정은 이제 현실에 가까워지고 있다.
스카이코비원·인보사·조인트스템의 사례는 각기 다른 문제를 비추는 것으로 보일 수 있으나, 그 근원은 동일하다. 한국의 규제기관은 그 본질적 책무인 '좋은 신약이 환자에게 빠르게 도달하게 돕는 것'에 있어 충분히 능동적이지 못하다. 스카이코비원이 빠르게 허가 받을 수 있도록 레퍼런스 시스템을 장착하지 못했고, 인보사가 과오를 딛고 부활할 수 있도록 도움을 주지 못했고, 조인트스템이 명징한 기준 하에 평가될 수 있는 시스템을 마련하지 못했다.
이는 FDA와 확연히 비교되는 자세이기도 하다. FDA는 항체, 케미컬, 펩타이드, ADC(항체약물접합체) 등이 해결해줄 수 없는 미충족 수요인 '첨단 재생 치료'에 대해 기민하게 대응하고 있다. 일례로 스트라타테크의 열화상 세포치료제인 '스트라타그래프트(STRATAGRAFT)'는 FDA로부터 RMAT(Regenerative Medicine Advanced Therapy·첨단재생의학치료제)으로 지정된 후, BLA(품목허가 신청서) 제출일로부터 1년만에 허가를 득했다. RMAT 지정을 통해 FDA 전문 인력의 자문을 받으며 '좋은 약이 올바른 방법으로 빠르게 환자에게 갈 수 있도록' 서포트를 받은 덕이다.

이러한 심사 기조, 즉 첨단 재생 치료제가 환자에게 빠르게 도달하게끔 도우려는 FDA의 의지는 나날이 강력해지고 있다. 이는 코로나19 백신의 초고속 허가를 지휘했던 피터 막스(Peter Marks) FDA CBER(생물의약품평가센터) 국장의 의지이기도 하다. 그의 지난 발언을 종합해 보건대, 막스 국장은 '좋은 약이 빠르게 환자에게 가도록 돕는다는 규제 기관의 본질적 의무를 지키면, 미국의 제약기업은 자연스럽게 시장을 지배해 국익에 보탬이 될 것'이라 여기는 것으로 보인다. 즉 막스 국장은 FDA를 '규제'기관이되 방관하지 않는 '지원'기관으로 이해하고 있다.
다시 한국으로 돌아오면, 우리 규제기관은 좋은 약을 구분하는 기능에서도, 좋은 약을 빠르게 환자에게 가져다주는 기능에서도 아쉬운 부분이 크다. 여기에 실망한 기업들은 결국 미국으로, 유럽으로, 중국으로 빠져나간다. 이런 현상이 지속되면 한국은 자국의 기업들에게 마저도 '코리아 패싱'을 당하고 만다. 자국의 기업이 개발한 신약이 자국민에게 가지 못하고, 타국에 먼저 가 버리는 불상사는 그리 먼 미래에 있지 않다.
그러나 한국 규제기관의 역량에 대한 지적은 이미 고루하다. 기계적인 비판은 아무런 가치도 만들어내지 못한다는 사실을 직시하고, 실천적인 해결책을 찾아 나서야 한다. 그 해결책은 결국 '인력의 질'을 상승시키는 데 초점을 둬야 할 것으로 보인다. FDA 심사관보다 몇 배나 많은 업무량과 책임을 지면서도, 몇 배나 낮은 보상을 받는 국내 심사관들의 사정을 재고해야 한다는 이야기다.
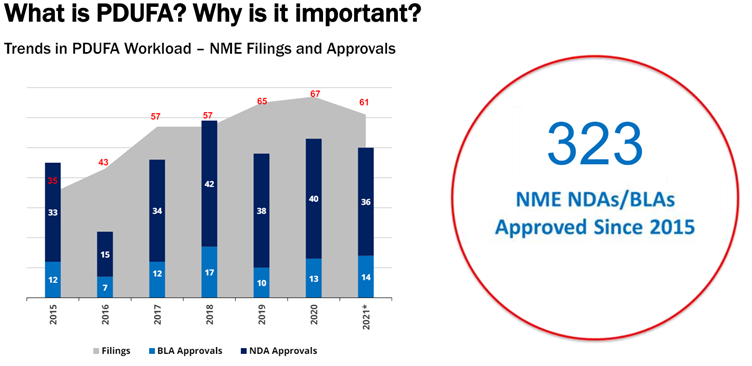
우리는 FDA도 같은 문제를 겪은 적이 있음을 기억해야 한다. 1990년대 이전의 FDA는 심사관의 숫자도, 전문성도 기대에 못 미치는 수준이었다. FDA는 이를 해결하고자 1992년에 '피두파(PDUFA·Prescription Drug User Fee Act)' 법을 제정해, 제약사들이 십시일반 모은 자금으로 고급 인력을 채용하고 심사관들을 교육했다. 문제가 인력이라는 것을 30년도 전에 파악해 해결에 나섰다는 부분에 눈길이 쏠린다.
PDUFA와 같은 법안을 우리도 제정할 수 있을지, 그것이 현실적으로 어렵다면 어떤 대안을 추진해야 할지 고민이 되는 지점이다. 문제는 정의됐다. 이제 우리에게 남은 건 해결책을 찾아 나서는 일이다. 쪼그라드는 내수 시장에, 본분을 다하지 못하는 규제 시스템에 허덕이는 기업들이 자국민마저 떠나는 현상이 본격화되기 전에 말이다.
히트뉴스(hitnews.co.kr) 박성수 기자 입력 2024.07.08 06:07