'세포·유전자 치료제' 쏟아지는데 MADE IN KOREA '실종'
조인호 범부처재생의료기술개발사업단 단장 "2019년 4월 이후 국내 제품 전무"

이는 조인호 범부처재생의료기술개발사업단 단장이 한국제약바이오협회가 발간한 'KPBMA FOCUS'를 통해 주장한 내용이다. 팜뉴스가 보고서에 담긴 조 단장의 목소리를 토대로, 업계가 곱씹을 만한 대목들을 재구성했다.
# 글로벌 시장, 세포·유전자 치료 신약 개발 각축전
글로벌 시장에서는 세포·유전자치료제 1개 제품에서 연간 1조원 이상 매출액이 나오는 제품들이 나오고 있다. 길리어드의 예스카타 CAR-T 세포·유전자치료제는 2023년 기준 연 매출 1조 원을 돌파했다.
Evaluate Pharma(글로벌 시장조사 전문기관)에 따르면, 재생의료 분야 중 세포·유전자치료제의 시장규모가 2021년부터 2026년까지 무려 연평균 49%의 높은 성장률을 보일 것으로 예측했다. 반면 합성의약품 성장률은 연평균 5.7% 성장에 그칠 것이라고 전했다.
글로벌 다국적 제약사 노바티스가 2017년 CAR-T 세포·유전자치료제인 킴리아를 개발한 이후 미국 및 EU를 중심으로 세포·유전자치료제는 전 세계적으로 빠르게 품목허가가 이뤄지고 있다.
특히 미국 식품의약국(FDA)은 2020년 이후 2024년 4월까지 총 20개 제품을 허가 하였으며, 최근 미국 FDA 심사 현황 자료에 의하면 2024년 5월 이후 연말까지 4개 제품의 추가 시판이 예상된다. 유럽의약품청(EMA)도 같은 기간 11개 제품을 허가했다.

# 미국·유럽 주력...관련 법 정비하고 신속 개발 지원
그렇다면 미국과 유럽이 글로벌 세포·유전자치료제 시장의 선두 주자로 등극한 이유는 뭘까.
FDA는 재생의료 첨단의약품에 해당하는 의약품이 신속심사제도를 이용할 수 있도록 다양한 제도를 운영하고 있다.
특히, 2016년 21세기 치료법(21st Century Cures Act)을 제정한 이후 새로운 치료법 개발과 첨단의약품 및 의료기기 개발 촉진을 위한 신속한 허가를 지원하고 있다.
동법을 통해 첨단재생의료치료제(Regenerative Medicine Advanced Therapy, RMAT)에 대한 정의와 범주를 신설하고 불필요한 규제들을 정비했다.

RMAT은 의학적 미충족 수요(Medical Unmet Needs)를 해결할 수 있는 재생의료치료제의 개발을 가속화하기 위한 제도다. RMAT 지정을 받게 되면 FDA가 시행 중인 신속 개발 프로그램의 혜택을 받을 수 있다.
최근 미국의 세포·유전자치료제 승인 건수 증가는 FDA의 혁신 신약 허가 지원 제도 도입 등 신약 개발에 대한 우호적인 분위기에 힘입은 결과다.
유럽도 다르지 않다. EU는 2007년 첨단의료제품(Advanced Therapy Medical Product, ATMP)의 개발과 시판 허가를 촉진하기 위하여 2007년 특별법을 제정했다.
이를 통해 EU 회원국 간의 제품의 안전성과 사후 추적관리를 효율적으로 관리하고 첨단의약품 분야의 전문성을 공유하기 위해 중앙허가제를 도입했다.
ATMP의 개발을 위한 임상시험 신청서는 국가 관할 당국에 개별적으로 제출되지만, 판매 승인의 경우 유럽의약품청(EMA)에 의해 EU 전역에 적용 가능한 중앙집중식 승인 및 허가를 받고 있다.
# 우리나라, '법'도 있고 '지원'도 했는데...실종?
우리나라도 2021년 첨단재생의료 및 첨단바이오의약품 심의위원회가 구성되고 사무국이 설치되어 ‘첨단재생의료 임상연구제도’를 운영 중이다. 2024년 3월까지 22건의 연구과제에 대해 총 167억 원의 임상연구비를 지원했다.
2021년 3월 과학기술정보통신부와 보건복지부가 공동 설립한 범부처재생의료기술개발사업단을 통해 2024년 6월 기준 173개 과제에 1,799억 원을 지원했다.
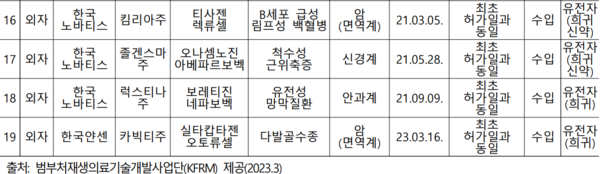
그런데도 2001년 이래 총 15개 품목의 세포치료제 제조허가 실적을 보유했을 뿐, 2019년 4월 이후 국내 제품은 단 한 건도 품목허가를 받은 제품이 없다. 4건의 수입 유전자치료제에 대한 품목허가 실적만 있다.(하단 표 참고)
물론 우리 정부도 첨단재생바이오법 제정(2019년), 개정(2024년)은 물론 최근에는 첨단바이오 이니셔티브를 발표하며 대응해왔다.
# MAIE IN KOREA? 신속 심사 제도 개선 급선무
그러나 국내 재생의료 생태계는 여전히 혁신기술 발굴, 투자유치, 인프라, 인허가 규제장벽 등의 문제를 안고 있어 세계시장 및 기술개발 발전 속도를 따라가지 못하고 있다.
특히 최근 많은 벤처, 중소기업들은 국내 시장에서의 투자 유치 및 임상시험 인허가의 현실적인 어려움으로 국외 기술이전 및 전략적 제휴 등을 통한 국외 진출을 모색 중이다.
국내 우수 핵심 기술이 싼값으로 해외로 유출될 가능성이 있으며, 향후 기술 종속국이 될 수 있어 대책 마련이 시급하다.
따라서 세포·유전자 치료제 등 첨단바이오분야는 혁신 도전적인 과제 발굴과 추진을 위해 범정부 차원의 지원이 필요하다.
국가 예산 배정은 물론 과제 기획과 선정, 관리는 빠르게 변화하는 글로벌 연구환경을 반영할 수 있도록 유연하게 적용할 필요가 있다.
무엇보다도 세포·유전자치료제의 인허가는 미국, 유럽, 일본 등 선진국의 품목허가 성공 사례를 분석해서 첨단재생의료 신기술의 빠른 확산을 위해 적합한 신속심사제도 마련해야 한다.
규제 허들을 넘을 수 있는 밀착지원 컨설팅 등 규제 및 지원체계를 조속히 재정비할 필요가 있다는 뜻이다.
마지막으로, 첨단재생바이오법이 개정되었으나 법 개정은 원론적인 내용인 만큼 국내 세포·유전자 치료제 개발이 가속화되기 위해서는 시행령에 구체적인 내용이 담겨야 하며, 향후 합리적인 규제 기준 마련이 절실하다.
팜뉴스(pharmnews.com) 최선재 기자 입력 2024.06.28 06:00